You may also like...
Boxing Giant Anthony Joshua Teases Explosive Crossover to MMA

Former heavyweight champion Anthony Joshua is contemplating a move into mixed martial arts, inspired by the trend of MMA...
Shocking Courtside Drama: Mystics Star Ejected, Escorted by Police

Washington Mystics coach Sydney Johnson was ejected from Saturday's game against the Atlanta Dream after a heated exchan...
Horror Reigns! 'Scary Movie' Shatters Records With $55M Opening While 'Masters of the Universe' Fails to Impress

Scary Movie has defied expectations by dominating the box office with a record-breaking opening, leading a surprising su...
Tribeca Festival Erupts: Outcry Over 'Dog Rape' Joke Sparks Furious Condemnation on Red Carpet!

The Tribeca Festival has vehemently condemned jokes made by Elon Gold and Lizzy Savetsky on the red carpet of “The Weddi...
An 'Old-School' Mystery: Lonely Grand Piano Captivates South Africa's Level Four
Level Four restaurant in Rosebank, Johannesburg, offers high standards and a familiar menu within a classy boutique hote...
Nostalgia Alert! Iconic 2000s Stores You Absolutely Missed

The American mall landscape is rapidly changing, leading to a nostalgic reflection on beloved retailers from the 2000s. ...
MTN's Tax Shock: Ghana Takes Heavier Toll Than Nigeria!
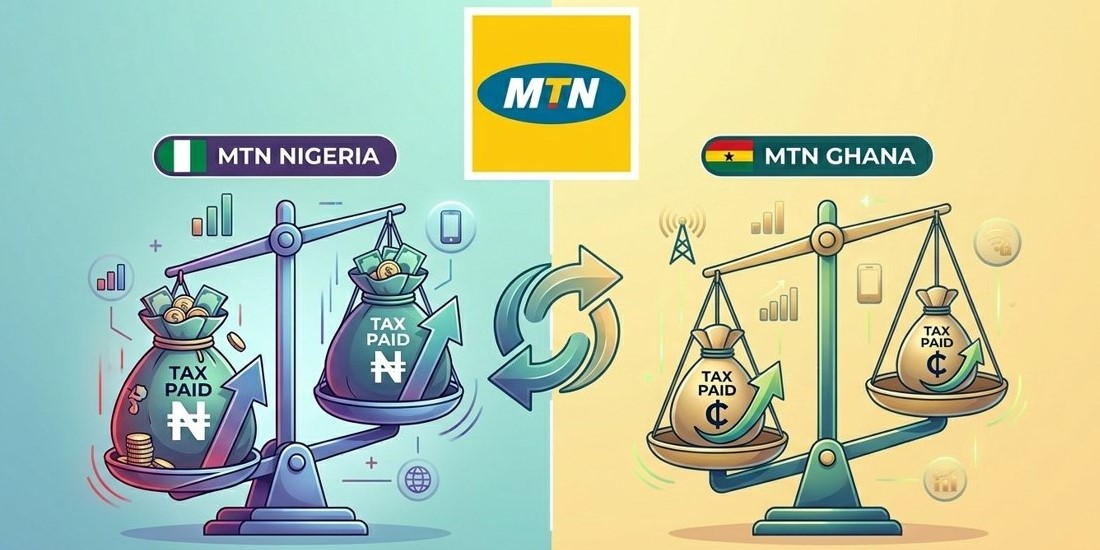
MTN Group reported a significant increase in tax payments to African governments in 2025, totaling R61.11 billion, drive...
Africa Hails New Tech Prodigy: Meet The Youngest Microsoft Specialist!

Six-year-old Damilare Akano has made history as Africa's youngest Microsoft specialist, achieving top honors in the Cert...